What is Phylogeny?
In general, phylogeny is the study of evolutionary development or history of a specific group of species, specifically a taxonomic group of organisms [1]. It can be applied to physical features or exact sequences such as those found in genes or proteins. Homologous sequences can be compared across genetically related organisms to discover their evolutionary relationships [2]. These relationships can then be used to construct phylogenetic trees.
Phylogenetic trees are commonly used for depicting evolutionary relationships among organisms. Figure 1 provides an example phylogenetic tree with labeled components. The root connects the original most common ancestor among all species. When the root undergoes a speciation event two daughter lineages are created. The daughter lineages continue to undergo speciation events over and over again producing nodes in the process. Nodes are points of common ancestors for the following branched species. External nodes or branch tips are the final taxa in the study. Organisms can be organized into a clade when they're believed to have evolved from a common ancestor. Clades can include living and extinct descendants of that common ancestor [3].
Phylogenetic trees are commonly used for depicting evolutionary relationships among organisms. Figure 1 provides an example phylogenetic tree with labeled components. The root connects the original most common ancestor among all species. When the root undergoes a speciation event two daughter lineages are created. The daughter lineages continue to undergo speciation events over and over again producing nodes in the process. Nodes are points of common ancestors for the following branched species. External nodes or branch tips are the final taxa in the study. Organisms can be organized into a clade when they're believed to have evolved from a common ancestor. Clades can include living and extinct descendants of that common ancestor [3].
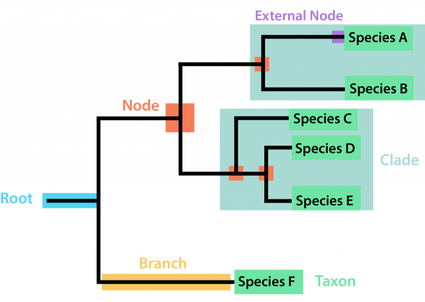
Figure 1. A sample phylogenetic tree with labeled key components.
Methods for Constructing Phylogenetic Trees
There are various methods that can be used when constructing a phylogenic tree. These methods can produce similar or different results due to their differing procedures explained below [4].
Maximum Likelihood Method (ML)
This method simultaneously evaluates all possible tree's and calculates the probabilities of each scenario. The Maximum Likelihood tree is selected as the one with the highest probability of occurring. This method is computationally intense, but often the most preferred and informative.
Neighbor Joining Method (NJ)
To begin, each pair is evaluated for being joined and the sum of all branch lengths of the resultant tree are calculated. The pair that yield the smallest sum is considered the closest neighbors and is thus joined. A new branch is built between the pair and the rest of the tree and branch lengths are recalculated. This process is repeated until one taxa remains. The NJ method is generally rapid and accurate, but only produces a single tree and doesn't consider other options.
Minimum Evolution Method (ME)
The ME method aims to create a tree with the minimum sum of branch lengths possible. It fixes the internal nodes by using the distance to external nodes and then optimizes the internal branch lengths to build a final tree.
Steps for Constructing a Phylogenetic Tree
1.) Compile Sequences
To begin, homologs desired to be included within the phylogenetic tree must be identified. Various sources such as the Homologene tool within GenBank can provide specific sequences of the desired gene or protein for homologs. Additionally, the general Blast tool within NCBI can be utilized across species. The sequences should be saved as a plain text file in FASTA format. Below you will find the SYNGAP1 protein sequences across homologs along with a downloadable file.
| |||
2.) Align Sequences
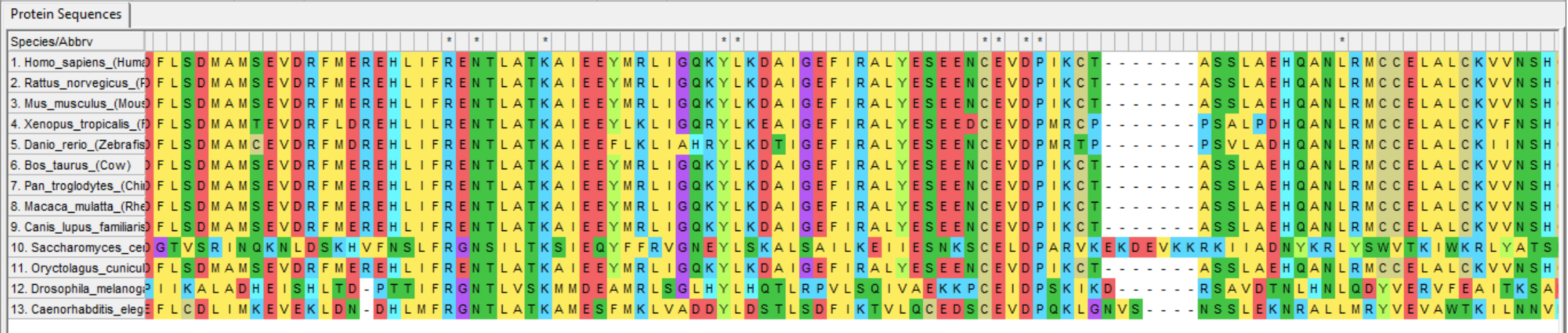
After the sequences have been collected they must be aligned for proper comparison. Sequences can be uploaded to ClustalOMEGA or MEGA for automatic alignment. Below is a small section of the SYNGAP1 protein sequence alignment. The organisms are indicated on the left and each letter resembles an amino acid. The top bar shows asterisks where the amino acid is conserved across all species.
3.) Construct Tree
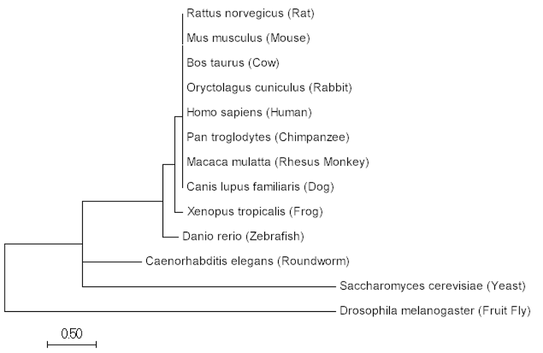
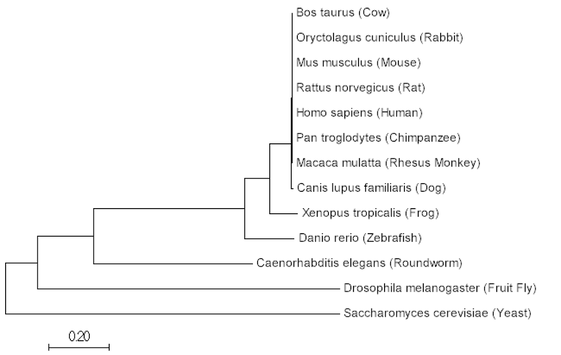
The aligned sequences can then be used to construct a tree within the same programs, ClustalOMEGA or MEGA. Below are phylogenetics tree's utilizing the Maximum Likelihood, Neighbor Joining and Minimum Evolution Method for SYNGAP1 protein.
Maximum Likelihood
Neighbor Joining Method
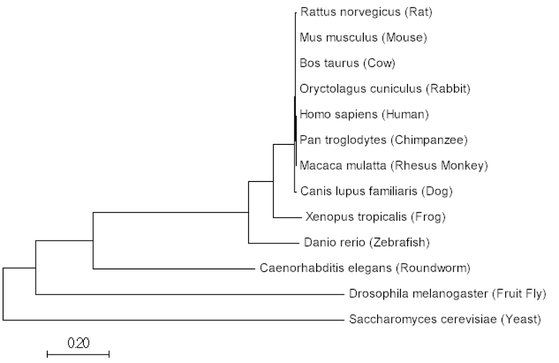
Minimum Evolution Tree
Discussion
All three methods produce extremely similar results for SYNGAP1 sequences. Considering, SYNGAP1 plays an important role in neuron function it makes sense that organisms have conserved this gene over time and have maintained a similar copy. The phylogenetic trees show similarity between related nervous systems. It is also interesting to see yeast within this tree and that it is the most distant from all other organisms. This makes sense due to the fact that yeast don't possess a nervous system; however, SYNGAP1 has GTPase activation properties that could play a role in yeast outside of the nervous system.
References
[1] "Phylogeny". Biology-Online Dictionary. (2014). Retrieved from https://www.biology-online.org/dictionary/Phylogeny
[2] Waikagul, Jitra, and Urusa Thaenkham. “Methods of Molecular Study: DNA Sequence and Phylogenetic Analyses.” Science Direct, Approaches to Research on the Systematics of Fish-Borne Trematodes, 10 Mar. 2014. Retrieved from www.sciencedirect.com/science/article/pii/B9780124077201000066.
[3] "Understanding phylogenies." Understanding Evolution. Berkeley. Retrieved from https://evolution.berkeley.edu/evolibrary/article/evo_05
[4] Li, Yan. How To Build a Phylogenetic Tree. guava.physics.uiuc.edu/~nigel/courses/598BIO/498BIOonline-essays/hw2/files/hw2_li.pdf.
Images and Figures:
Header: https://dumielauxepices.net/wallpaper-637799
Figure 1: http://library.open.oregonstate.edu/microbiology/chapter/taxonomy-evolution/
Alignment and Phylogenetic Trees created by Abigail Jaquish
[2] Waikagul, Jitra, and Urusa Thaenkham. “Methods of Molecular Study: DNA Sequence and Phylogenetic Analyses.” Science Direct, Approaches to Research on the Systematics of Fish-Borne Trematodes, 10 Mar. 2014. Retrieved from www.sciencedirect.com/science/article/pii/B9780124077201000066.
[3] "Understanding phylogenies." Understanding Evolution. Berkeley. Retrieved from https://evolution.berkeley.edu/evolibrary/article/evo_05
[4] Li, Yan. How To Build a Phylogenetic Tree. guava.physics.uiuc.edu/~nigel/courses/598BIO/498BIOonline-essays/hw2/files/hw2_li.pdf.
Images and Figures:
Header: https://dumielauxepices.net/wallpaper-637799
Figure 1: http://library.open.oregonstate.edu/microbiology/chapter/taxonomy-evolution/
Alignment and Phylogenetic Trees created by Abigail Jaquish
This webpage was produced as an assignment for Genetics 564, an undergraduate capstone at University of Wisconsin- Madison.
